Повне екзомне секвенування (WES) — це сучасний і максимально інформативний метод генетичної діагностики.
Воно дозволяє одночасно проаналізувати всі білок-кодуючі ділянки геному людини — близько 20 000 генів,
де зосереджено до 85 % усіх відомих патогенних мутацій.
Що таке екзом
Екзом — це сукупність ділянок ДНК, які містять інформацію для синтезу білків.
Хоча ці ділянки становлять лише близько 1 % усього геному, саме тут виникає більшість генетичних змін,
що призводять до спадкових захворювань.
Що досліджує повне екзомне секвенування (WES)
Повне екзомне секвенування дозволяє виявляти широкий спектр змін у ДНК, зокрема:
- одиночні нуклеотидні варіанти (SNV) — заміни однієї «літери» ДНК;
- невеликі вставки та делеції (indel);
- більші делеції та дуплікації (CNV);
- а також варіанти в мітохондріальному геномі, який також секвенується повністю.
Таким чином, WES охоплює як ядерний, так і мітохондріальний геном, що робить дослідження максимально інформативним і клінічно значущим.
Коли показане повне екзомне секвенування
- за складних або неспецифічних симптомів, коли важко визначити причину захворювання;
- при підозрі на генетично гетерогенне захворювання (коли різні гени можуть спричиняти схожі прояви);
- якщо цільові генетичні тести недоступні або дали негативний результат;
- за необхідності повторного аналізу раніше невизначених випадків.
Технічні характеристики методу
Для аналізу використовується технологія повного захоплення екзому та високопродуктивне секвенування нового покоління (NGS)
на платформах Illumina NovaSeq / NextSeq (paired-end, 2×150 bp).
Усі етапи виконуються в лабораторії, акредитованій за стандартами CLIA, CAP та ISO 15189,
що гарантує клінічний рівень якості даних.
- Метод збагачення: повний екзомний захоплення, включно з UTR та мітохондріальним геномом
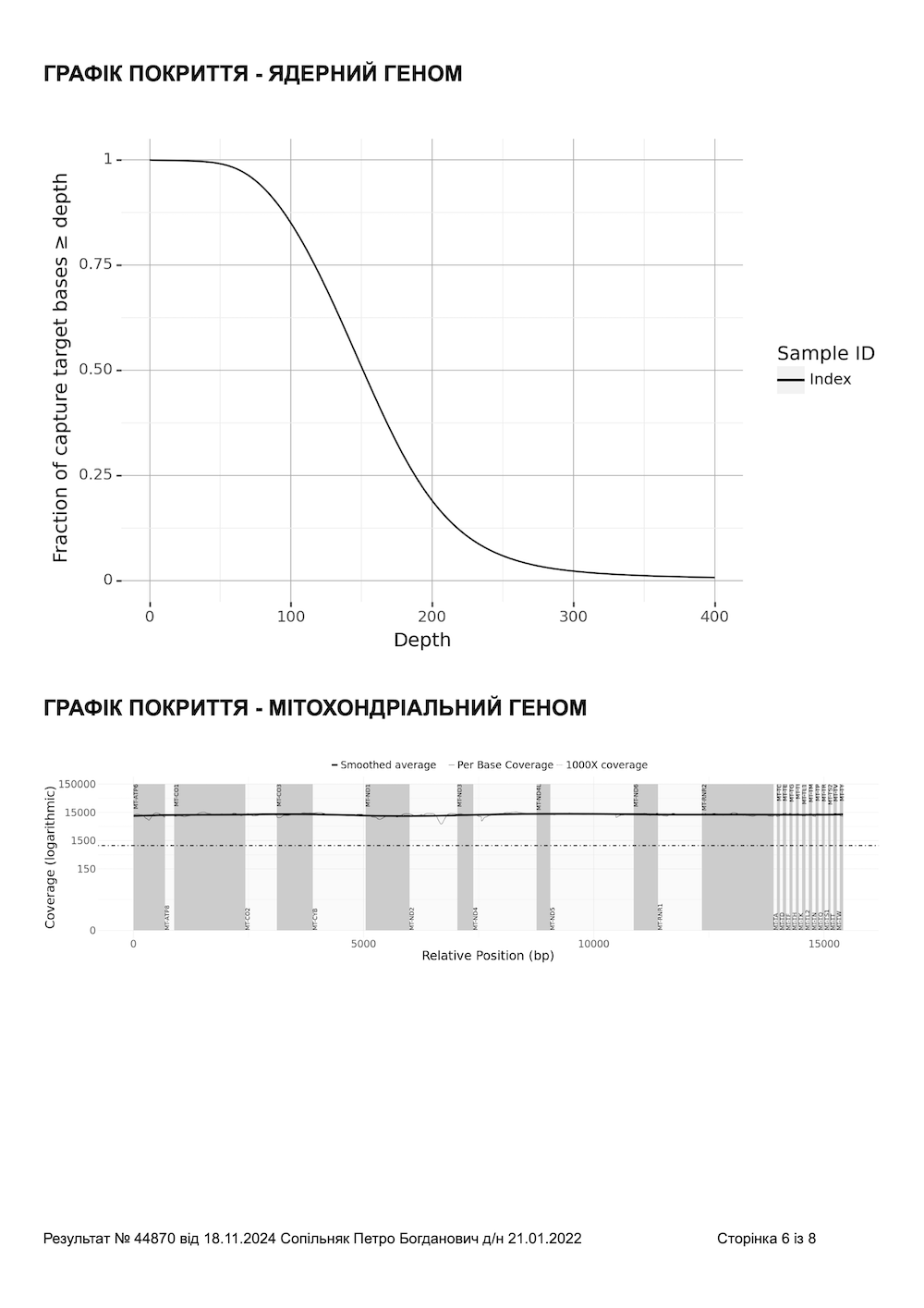
- Середнє покриття екзому: 150× (у середньому 140–180×)
- Покриття ≥20×: понад 99 % екзомних ділянок
- Чутливість SNV: 99,7 %, специфічність >99,99 %
- Чутливість indel: 97,0 %, специфічність >99,99 %
- Виявлення CNV: висока точність для делецій і дуплікацій (чутливість >92 % для 1 екзону, >98 % для ≥5 екзонів)
- Точність аналізу: відтворюваність та повторюваність — 99,7 %
- Мітохондріальний геном: секвенується повністю з глибиною покриття >1000× і охопленням ≥99,9 % — включено в кожне дослідження WES
- Обробка даних: вирівнювання прочитань до еталонного геному та пошук змін (варіантів) із використанням перевірених алгоритмів клінічного рівня
- Інтерпретація: кожна знайдена заміна або делеція проходить фільтрацію та перевірку за міжнародними базами даних — ClinVar, gnomAD, OMIM, HGMD та іншими
- Класифікація варіантів: проводиться за критеріями Американського коледжу медичної генетики (ACMG, 2015) з поділом на категорії: патогенний, ймовірно патогенний, варіант із невизначеним значенням, ймовірно доброякісний і доброякісний
- Надання даних: файли BAM і VCF доступні за запитом
Клінічний аналіз та інтерпретація
Після секвенування дані проходять багаторівневу біоінформатичну обробку.
Спеціалізовані алгоритми порівнюють мільйони прочитань із еталонним геномом, виділяючи клінічно значущі варіанти.
Кожен із них оцінюється та класифікується за міжнародними критеріями ACMG (2015).
У звіт включаються лише ті зміни, які мають клінічне значення для конкретного пацієнта. Це можуть бути:
- гетерозиготні варіанти при аутосомно-домінантних захворюваннях;
- гомозиготні або складні гетерозиготні варіанти при аутосомно-рецесивних захворюваннях;
- гемізиготні варіанти при Х-зчеплених захворюваннях.
Обмеження методу
Метод не виявляє:
- збалансовані транслокації та інверсії;
- генні конверсії;
- експансії повторів (наприклад, хвороби з групи тринуклеотидних повторів);
- варіанти поза межами ±20 пар нуклеотидів від екзон-інтронної межі;
- структурні перебудови без зміни кількості копій.
Метод може мати знижену чутливість при:
- низькому рівні мозаїцизму (частка мутантного алеля < 15 %);
- наявності псевдогенів або дубльованих ділянок;
- інсерціях або делеціях довжиною понад 50 пар нуклеотидів;
- варіантах у сегментально дубльованих областях геному.
Що відбувається після тесту
Якщо виявлено патогенний варіант, його можна підтвердити методом
Сенгера у родичів для уточнення типу успадкування.
Якщо змін не виявлено, це не виключає генетичну природу захворювання —
деякі варіанти ще не описані в науковій літературі або не визначаються цим методом.
Пацієнт може замовити перегляд даних через 12–24 місяці.
Під час перегляду враховуються оновлені бази даних і нові наукові публікації.
Лабораторія надає один безкоштовний перегляд (для варіантів, зазначених у попередньому звіті)
та повну послугу повторного аналізу з новим секвенуванням за потреби.
Переваги WES у нашій лабораторії
- Міжнародне визнання та акредитації. Аналіз виконується в міжнародній лабораторії Blueprint Genetics, що входить до корпорації Quest Diagnostics (США). Лабораторія сертифікована за стандартами CLIA, CAP та ISO 15189, що гарантує клінічний рівень якості на кожному етапі — від виділення ДНК до інтерпретації результатів.
- Повне охоплення екзому та мітохондріального геному. Аналіз включає всі білок-кодуючі гени людини (≈ 20 000) і повний мітохондріальний геном, забезпечуючи максимальну діагностичну інформативність.
- Виявлення не лише точкових, а й великих змін. Тест визначає однонуклеотидні варіанти, невеликі вставки та делеції, а також делеції й дуплікації (CNV) по всьому екзому — з високою чутливістю навіть для окремих екзонів.
- Розширене охоплення геному. Окрім кодуючих ділянок, аналіз включає близько 1500 клінічно значущих некодуючих варіантів, які можуть спричиняти захворювання і зазвичай не входять до стандартних панелей інших лабораторій.
- Передові технології секвенування. Використовуються платформи Illumina NovaSeq / NextSeq та методи глибокого секвенування (середнє покриття ≈ 150×), що забезпечує рівномірне покриття всіх генів і виняткову точність даних.
- Точна клінічна інтерпретація. На відміну від багатьох лабораторій, ми не надаємо перелік усіх знайдених змін.
У фінальному звіті подаються лише клінічно значущі варіанти, що можуть бути пов’язані із симптомами пацієнта.
Для кожного варіанта додається розгорнутий клінічний опис — чому він може (або не може) пояснювати спостережуваний фенотип,
із аналізом усіх доступних наукових публікацій та даних із міжнародних баз.
- Власний біоінформатичний аналіз і контроль якості. Blueprint Genetics застосовує удосконалений програмний конвеєр аналізу та багаторівневу систему перевірки, що забезпечує високу точність і відтворюваність результатів.
- Розпізнавання рідкісних і некодуючих варіантів. У дослідження включено близько 1500 відомих некодуючих патогенних варіантів, які часто не аналізуються в інших лабораторіях.
- Прозорість і можливість повторного перегляду. За бажанням можна отримати вихідні дані секвенування (.BAM, .VCF) і замовити безкоштовний перегляд результатів у разі появи нових наукових даних.
Що далі?
Якщо аналіз WES виявив варіанти, що можуть бути причиною захворювання, можна перевірити їх наявність у батьків. Аналіз окремих мутацій методом Сенгера.
Якщо аналіз не виявив варіантів, це може означати, що науці поки невідомий цей варіант або його неможливо визначити методом WES (NGS другого покоління).
Можна отримати сирі дані і проводити повторний перегляд кожні кілька років — із часом можуть бути відкриті нові генетичні чинники хвороби.
Лабораторія надає один безкоштовний перегляд даних, рекомендовано замовляти його через 12 місяців або за появи нових симптомів, але не пізніше 24 місяців від дати отримання результату.
У межах цієї опції переглядаються лише варіанти, зазначені в попередньому звіті.
Також лабораторія пропонує повторний аналіз даних — у цьому випадку зразок секвенується повторно і проходить повний аналіз.
Приклад результату WES
Клінічна історія
Хворий хлопчик 2 років із підозрою на аутизм, затримкою дрібної моторики, затримкою мовлення, гіпертонусом і двобічним крипторхізмом. Не розмовляє майже 2 роки, погано спить, не реагує на ім’я. Не жує, їсть лише подрібнену блендером їжу. Має довгий фільтр, загнуті ніздрі, гіпертрихоз, плоскостопість. В родинному анамнезі подібних випадків немає. Раніше проведені генетичні дослідження (каріотип 46, XY, тест на синдром Сміта-Магеніса) — без патологічних змін.
Результат повного екзомного секвенування
Аналіз повного екзому виявив гетерозиготний варіант ANKRD11 c.1903_1907del, p.(Lys635Glnfs*26). Цей варіант класифіковано як патогенний на основі наявних даних, що підтверджують його хвороботворну роль. Захворювання, спричинене варіантами ANKRD11, успадковується за аутосомно-домінантним типом і зазвичай виникає як варіант de novo. Рекомендовано консультацію генетика та тестування членів родини.
Як підготуватися
Кров можна здавати не натще.
Трансплантація кісткового мозку є протипоказанням.
У разі переливання крові забір слід проводити не раніше ніж через 3 тижні.
Необхідно надати висновок лікаря з описом симптомів і попереднім діагнозом, бажано — фото пацієнта (якщо є фенотипові особливості), а також заповнити електронну анкету.
Після отримання результатів обов’язкова консультація лікаря-генетика для інтерпретації звіту та визначення подальших дій.
Додаткова інформація

 Київ
Київ