В панель "Краніосиностоз" входить 38 генів, також включена оцінка некодуючих варіантів, виявлення делецій та дуплікацій.
Ідеальна панель для пацієнтів із клінічною підозрою на краніосиностоз.
Краніосиностоз визначається як передчасне зрощення одного або кількох черепних швів, що призводить до вторинного спотворення форми черепа. Це може бути наслідком первинного дефекту окостеніння (первинний краніосиностоз) або частіше порушення росту головного мозку (вторинний краніосиностоз). Передчасне закриття швів призводить до збільшення тиску всередині голови та деформації зовнішнього вигляду черепа та лицьових кісток з різним проявом.
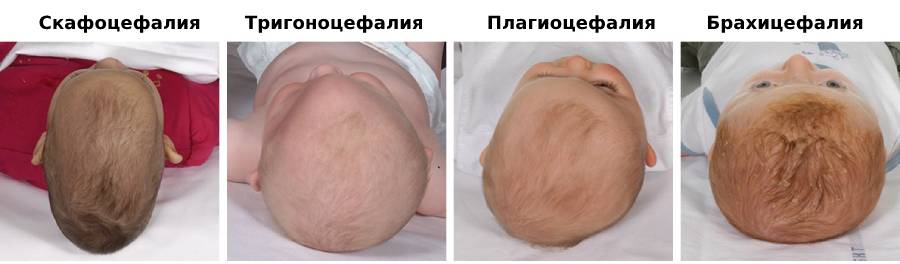
Залежно від того, які черепні шви зростаються раніше, череп може мати різну форму:
скафоцефалія, плагіоцефалія, тригоноцефалія, брахіцефалія.
Основною причиною розвитку краніосиностозу є генетичні порушення.
Краніосиностоз може виникати ізольовано або як частина синдрому з різними патернами наслідування та ризиком рецидиву.
Краніосиностоз зустрічається у 1 із 2200 новонароджених.
Гени, що входять до панелі:
| Ген |
Асоційований фенотип |
Спадкоємність.* |
ClinVar** |
HGMD** |
|
ALPL
|
Одонтогіпофосфатазія, гіпофосфатазія перинатальна летальна, інфантильна, ювенільна та доросла форми |
AD/AR |
78 |
291 |
|
ALX3
|
Лобно-носова дисплазія 1 типу |
AR |
8 |
8 |
|
ALX4
|
Лобно-носова дисплазія 2 типу, тім'яні отвори |
AD/AR |
15 |
24 |
|
BMP4
|
Мікрофтальм, синдромальний, орофаціальна ущелина |
AD |
8 |
39 |
|
CDC45
|
Синдром Меєра-Горліна 7 |
AR |
10 |
19 |
|
EDNRB
|
Хвороба Гіршпрунга, синдром ABCD, синдром Ваарденбурга |
AD/AR |
12 |
66 |
|
EFNB1
|
Краніофронтоназальна дисплазія |
XL |
28 |
116 |
|
ERF
|
Краніосиностоз 4 |
AD |
17 |
16 |
|
ESCO2
|
Синдром СК фокомелії, синдром Робертса |
AR |
30 |
31 |
|
FGFR1
|
синдром Пфайффера, тригоноцефалія, гіпогонадотропний гіпогонадизм, остеоглофонічна карликовість - краніостеноз, синдром Хартсфілда |
AD/Digenic/Multigenic |
72 |
257 |
|
FGFR2
|
Синдром Аперта, синдром Пфайффера, синдром Джексона-Вейсса, лакримо-аурикулоденто-дигітальний синдром, синдром Бере-Стивенсона, синдром звивистих шкірних покривів, синдром Антлі-Бікслера без генітальних аномалій або порушень стероїдогенези Крузона, дисплазія вигнутої кістки |
AD |
100 |
154 |
|
FGFR3
|
Лакрімоаурікулодентодигітальний синдром, синдром Мюнке, синдром Крузона з чорним акантозом, камптодактилія, синдром високого росту та втрати слуху (CATSHL), ахондроплазія, гіпохондроплазія, танатофорна дисплазія 1 типу, танатофорна дисплазія 2
| AD/AR |
54 |
77 |
|
FLNB
|
Синдром Ларсена (домінантний), Ателостеогенез 1 типу, Ателостеогенез 3 типу, Спондило-карпало-тарзальна диспазія, Дисплазія бумеранга |
AD/AR |
43 |
121 |
|
FREM1
|
Роздвоєння носа, Манітобський окулотрихоанальний синдром, Тригоноцефалія |
AD/AR |
14 |
35 |
|
GDF5
|
Синдром множинних синостозів, малогомілкова гіпоплазія та складна брахідактилія типу А2
| AD/AR |
23 |
53 |
|
GLI3
|
Акрокалезний синдром, синдром Палістера-Холла, синдром цефалополісндактилії Грига, постаксіальна полідактилія типу А, преаксіальна полідактилія 3 типу, преаксіальна полідактилія 4 типу |
AD |
70 |
235 |
|
IFT122
|
Синдром Сенсенбреннера, Краніоектодермальна дисплазія (Левіна-Сенсенбреннера) 1 типу, Краніоектодермальна дисплазія (Левіна-Сенсенбреннера) 2 типу |
AR |
13 |
23 |
|
IFT140
|
Короткоберна грудна дисплазія з полідактилією або без неї, Асфіксуюча грудна дисплазія (ATD; Jeune) |
AR |
38 |
63 |
|
IL11RA
|
Краніосиностоз та зубні аномалії |
AR |
7 |
21 |
|
MASP1
|
синдром 3МС |
AR |
11 |
22 |
|
MEGF8
|
Синдром Карпентера 2 |
AR |
6 |
14 |
|
MSX2
|
Парієтальні отвори, Парієтальні отвори з ключично-черепною дисплазією, Краніосиностоз бостонського типу |
AD |
9 |
25 |
|
NOG
|
Синдром передплюсне-зап'ясткової коаліції, синдром множинних синостозів, анкілоз стремена з широкими пальцями рук і ніг (синдром Тейніссена-Кремерса), симфалангізм, проксимальний, брахідактилія типу B2 |
AD |
20 |
63 |
|
PAX3
|
Синдром черепно-лицьової глухоти, синдром Ваарденбурга |
AD/AR |
54 |
149 |
|
POR
|
Порушення стероїдогенезу через дефіцит цитохрому р450 оксидоредуктази, синдром Антлі-Бікслера |
AR |
14 |
70 |
|
RAB23
|
Синдром Карпентера 1 |
AR |
5 |
15 |
|
RECQL4
|
синдром Баллера-Герольда, синдром РАПАДІЛІНО, синдром Ротмунда-Томсона |
AR |
82 |
114 |
|
SKI
|
синдром Шпрінцен-Гольдберга |
AD |
20 |
23 |
|
SOX10
|
Периферична демієлінізуюча невропатія, центральна дисмієлінізація, синдром Ваарденбурга та хвороба Гіршпрунга, синдром Каллмана |
AD |
56 |
148 |
|
SPECC1L
|
Ущелина особи, коса, 1, синдром Opitz GBBB, тип II |
AD |
7 |
8 |
|
TCF12
|
Краніосиностоз |
AD |
23 |
56 |
|
TGFBR1
|
Синдром Лойса-Дитца |
AD |
40 |
69 |
|
TGFBR2
|
Синдром Лойса-Дитца |
AD |
58 |
139 |
|
TWIST1
|
синдром Сетре-Шотцена, синдром Робінова-Сорауфа, краніосиностоз |
AD |
28 |
205 |
|
TWIST2
|
Синдром аблефарон-макростомії, синдром Барбера-Сея, осередкова лицьова шкірна дисплазія 3, тип Сетлейса |
AD/AR |
7 |
9 |
|
WDR19
|
Пігментний ретиніт, нефронофтиз, короткореберна грудна дисплазія з полідактилією або без неї, синдром Сеніора-Локена, краніоектодермальна дисплазія (Левіна-Сенсенбреннера) 1 типу, краніоектодермальна дисплазія (Левіна-Сенсенбре) (ATD; Jeune) |
AR |
33 |
43 |
|
WDR35
|
Краніоектодермальна дисплазія (Левіна-Сенсенбреннера) 1 типу, Краніоектодермальна дисплазія (Левіна-Сенсенбреннера) 2 типу, Синдром короткореберної полідактилії 5 типу |
AR |
28 |
31 |
|
ZIC1
|
Краніосиностоз 6 |
AD |
5 |
9 |
* Тип успадкування: аутосомно-домінантний (AD), аутосомно-рецесивний (AR), мітохондріальний (mi), Х-зчеплений (XL), Х-зчеплений домінантний (XLD) та Х-зчеплений рецесивний (XLR);
** ClinVar, HGMD - кількість варіантів гена, класифікованих як патогенні або ймовірно патогенні в базі даних ClinVar, HGMD.
Можлива персоналізація панелі
В панель дозволяється додати додаткові гени (до 200), пов'язані з симптомами, та видалити будь-які гени (щоб залишилося не менше 2). Вартість панелі може змінюватися, а саме:
- мала панель 2-25 генів ,
- середня панель 26-125 генів ,
- велика панель понад 126 генів .
Переваги цього тесту
- Лабораторія (США), акредитована САР
- Персонал, сертифікований CLIA, проводить клінічні випробування у лабораторії, сертифікованій CLIA.
- Потужні технології секвенування, передові методи збагачення мішеней та біоінформатичні пайплайни забезпечують точну аналітичну продуктивність.
- Ретельне складання клінічно ефективних та науково обґрунтованих генних панелей
- Аналіз некодуючих варіантів, що викликають захворювання
- Строга схема класифікації варіантів
- Систематичний робочий процес клінічної інтерпретації з використанням пропрієтарного програмного забезпечення, що забезпечує точну та простежувану обробку даних NGS.
- Докладний клінічний висновок
- Можна замовити сирі дані секвенування в форматі.vcf
Що далі?
Якщо аналіз виявив варіанти, що є можливою причиною хвороби, можна перевірити наявність цих варіантів у батьків. Аналіз окремих мутацій по Сенгеру.
Якщо аналіз не виявив варіантів, які є можливою причиною хвороби, можна розширити тестування до повного екзома Розширення панелі до повного екзома. Крім того, можна отримати сирі дані і робити повторну оцінку кожні кілька років. Можливо, згодом науці стануть відомі нові генетичні чинники захворювання.
Як пройти дослідження
Натще не потрібно, рясно пити воду.
Проводиться забір венозної крові в пробірку з ЕДТА 4 мл.
Забір матеріалу у нас у центрі або можна передати нам пробірку ЕДТА, взяту у будь-якій лабораторії. Про можливість забору у дітей уточнювати додатково.
У наших пунктах забору крім Києва забор матеріалу коштує .
Необхідно надати висновок лікаря з клінічною картиною (опис симтомів, діагноз), заповнити в електронному вигляді анкету.
Аналіз не проводиться для здорових людей без симптомів, у цьому випадку рекомендуються скринінгові аналізи на носійство.
За результатами секвенування необхідно отримати консультацію лікаря-генетика.

 Київ
Київ
