Що таке сирі дані?
Сирі дані секвенування зазвичай є ресурсом для вчених, які мають навички біоінформатики. Сирі дані секвенування вимагають спеціальних біоінформатичних інструментів для відкриття файлів та їх подальшого аналізу чи візуалізації. Ми не надаємо допомоги в інтерпретації та обробці сирих даних.
Умови отримання сирих даних
5 тижнів з моменту отримання результату секвенування або з моменту отримання заявки.
Можна замовити протягом 2 років після дати отримання результату.
Формати сирих даних
Для повноекзомного секвенування (WES) надаються файли .VCF, .BAM. Для панелей - файл .VCF.
BAM (Карта вирівнювання двійкових послідовностей) — це стандартний формат файлу, який містить зчитування послідовностей, зіставлені з еталонним геномом людини, включаючи базову якість та показники якості зіставлення. Розмір файлів BAM становить ~6–10 ГБ для аналізу типу WES.
VCF (формат варіантів) — це стандартний формат файлу, в якому зберігається список варіантів послідовностей та їх геномних положень. Файл VCF не містить докладних інструкцій варіантів, таких як назва гена або частота популяції варіанту. Цей VCF не піддавався фільтрації відповідно до будь-яких конкретних стандартів якості. Його розмір становить ~2 МБ.
Що мені слід очікувати під час завантаження файлів?
Розмір файлу може бути більшим, тому ми рекомендуємо зарезервувати кілька годин на завантаження. Для завантаження файлів можна використовувати веб-браузер, але ми рекомендуємо використовувати утиліту командного рядка для завантаження файлів на комп'ютер, здатний зберігати і обробляти великі обсяги даних.
Ось так приблизно виглядають сирі дані після завантаження та відкриття текстовим редактором.
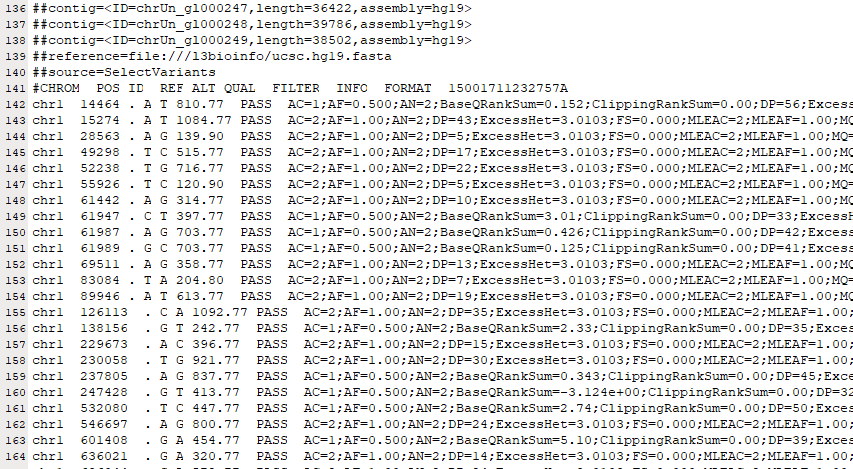
- Розмір файлу VCF (~2 МБ, WES та 400 МБ, WGS) зазвичай не становить проблем для звичайного комп'ютера. Для обробки файлів VCF потрібні біоінформаційні інструменти. Однак його можна відкрити в будь-якому текстовому редакторі або Excel після розпакування файлу VCF у форматі gz.
- Для файлів BAM (~6–10 ГБ, WES) важливо використовувати комп'ютер, який може зберігати та обробляти великі обсяги даних. Можна використовувати біоінформаційні інструменти для доступу до файлу BAM та виконання дзвінків варіантів (які створюють VCF) або візуалізації зчитувань та варіантів послідовностей.
Яке програмне забезпечення мені потрібне для читання сирих даних секвенування?
Аналіз сирих даних про послідовності вимагає знань та програмного забезпечення в галузі біоінформатики.
Для анотації та аналізу варіантів послідовності у файлі VCF доступно кілька комерційних та некомерційних інструментів. До найчастіше використовуваних і вільно доступних інструментів командного рядка належать:
- VEP (https://www.ensembl.org/info/docs/tools/vep/index.html)
- ANNOVAR (http://annovar.openbioinformatics.org/en/latest/)
- SnpEff (http://snpeff.sourceforge.net)
Існує також кілька некомерційних та комерційних програм на базі веб-браузера або автономних програм для анотування та аналізу варіантів. Для аналізу читання послідовностей GATK є одним із найчастіше використовуваних наборів інструментів [https://software.broadinstitute.org/gatk/].
Для візуалізації вмісту файлів VCF або BAM у геномному контексті можна використовувати Integrative Genome Browser (IGV) як автономну програму, так і інтерфейс веб-браузера.

 Київ
Київ
